
Frontotemporal Dementia

Frontotemporal dementia (FTD) describes a group of diseases characterized by degeneration of nerve cells - especially those in the frontal and temporal lobes of the brain. Unlike Alzheimer's disease, FTD usually does not include formation of amyloid plaques. In approximately 50% of people with FTD, there is an abnormal form of tau protein in the brain and about 50% of people with FTD have TDP-43 protein accumulation. A small percentage, about 5%, have FUS protein accumulation. This disrupts normal cell activities and may cause the cells to die.
Experts believe FTD accounts for 2 to 10 percent of all cases of dementia. Symptoms of FTD usually appear between the ages of 40 and 65. In many cases, people with FTD have a family history of dementia, suggesting that there is a strong genetic factor in the disease. The duration of FTD varies, with some patients declining rapidly over 2 to 3 years and others showing only minimal changes for many years. People with FTD live with the disease for an average of 5 to 10 years after diagnosis.
Because structures found in the frontal and temporal lobes of the brain control judgment and social behavior, people with FTD often have problems maintaining normal interactions and following social conventions. They may steal or exhibit impolite and socially inappropriate behavior, and they may neglect their normal responsibilities. Other common symptoms include loss of speech and language, compulsive or repetitive behavior, increased appetite, and motor problems such as stiffness and balance problems. Memory loss also may occur, although it typically appears late in the disease.
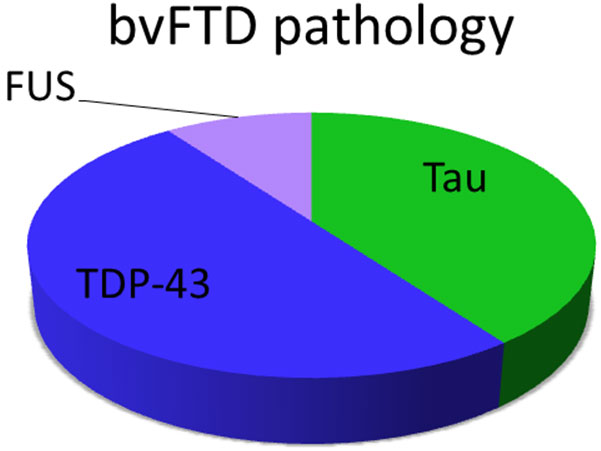
Proteins Associated with Behavioral Variant Frontotemporal Dementia (FTD)
Three types of proteins are associated with FTD: Tau, TDP-43, and FUS.
In one type of FTD called Pick's disease, certain nerve cells become abnormal and swollen before they die. These swollen, or ballooned, neurons are one hallmark of the disease. The brains of people with Pick's disease also have abnormal structures called Pick bodies, composed largely of the protein tau, inside the neurons. The cause of Pick's disease is unknown, but it runs in some families and thus it is probably due at least in part to a faulty gene or genes. The disease usually begins after age 50 and causes changes in personality and behavior that gradually worsen over time.
The symptoms of Pick's disease may include inappropriate social behavior, loss of mental flexibility, language problems, and difficulty with thinking and concentration. There is currently no way to slow the progressive degeneration found in Pick's disease. However, medication may be helpful in reducing aggression and other behavioral problems, and in treating depression.
In some cases, familial FTD is linked to a mutation in a gene called C9ORF72. This mutation is thought to be the most common cause of familial FTD and familial ALS (amyotrophic lateral sclerosis or Lou Gherig's disease). This discovery was made in 2011 and research is rapidly expanding to better understand the mechanism behind the genetic mutation, the relationship between the diseases, and possible treatment trials. A mutation in the tau gene (MAPT) or progranulin gene can also cause familial FTD.
Primary progressive aphasia (PPA) is progressive language disorder that causes a decline in functional ability. Symptoms may begin in people as early as their forties. "Aphasia" is a general term used to refer to deficits in language functions, such as speaking, understanding what others are saying, and naming common objects. In PPA one or more of these functions can become impaired. Symptoms often begin gradually and progress slowly over a period of years. As the disease progresses, memory and attention may also be impaired and patients may show personality and behavior changes. Many, but not all, people with PPA eventually develop symptoms of dementia.
There are 3 clinical types of PPA: nonfluent variant PPA (nfvPPA), semantic variant (svPPA), and logopenic variant (lvPPA). Patients with nfvPPA have problems with grammar, syntax, or speech production. Patients with svPPA have problems with word knowledge (semantics) and may have problems naming things. Both the nfvPPA and svPPA are typically associated with proteins that are seen in FTD. Patients with lvPPA may have problems with word finding or naming, but have intact word knowledge. The proteins seen in this disease are commonly the same proteins as seen in Alzheimer's disease. Some people believe that lvPPA is a type of Alzheimer’s disease.
Learn more about frontotemporal dementia.
Condition Spotlight




Clinical Trials
Clinical trials are research studies that evaluate a new medical approach, device, drug, or other treatment. As a Stanford Health Care patient, you may have access to the latest, advanced clinical trials.
Open trials refer to studies currently accepting participants. Closed trials are not currently enrolling, but may open in the future.